Chapitre 24
METHODES EN IMMUNOLOGIE
II – Mesure et
utilisation des anticorps
A – Mesure
du titre d’un anticorps.
A – 1 – Définitions
La présence d’un anticorps spécifique peut être mesurée par
plusieurs méthodes différentes. Certaines d’entre elles mesurent la fixation
directe de l’anticorps (Ac) sur l’antigène (Ag). Ces techniques sont basées sur
des interactions primaires alors que d’autres détectent les changements
physiques de l’antigène induit après la fixation de l’antigène et sont donc
basées sur des interactions secondaires. Quoi qu’il en soit, ces deux types de
tests peuvent être employés pour mesurer le titre et la spécificité des
anticorps produits au cours de la réponse immunitaire. Ces tests ayant été
utilisés historiquement pour détecter la présence d’anticorps dans le sérum de
patients, on les appellent communément tests sérologiques. La quantité
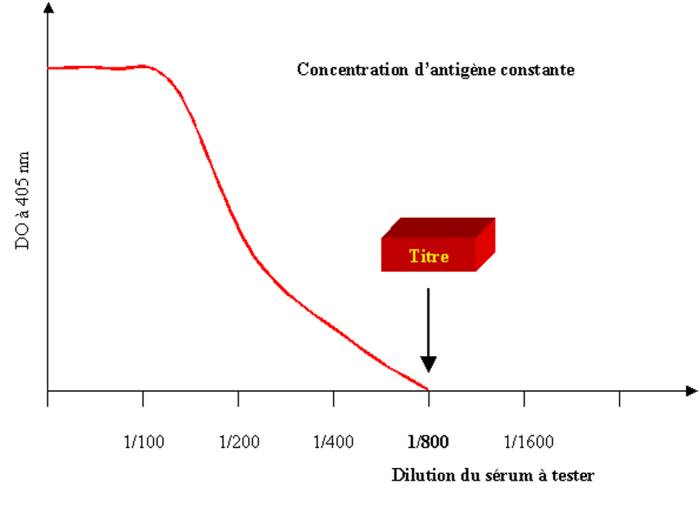
d’anticorps présente dans le sérum est déterminée par titrage de celui-ci en
dilution limite. Le titre d’un sérum correspond à l’inverse de la dernière
dilution positive. Il est à noter que certains auteurs définissent le titre
d’un sérum comme la dilution donnant 50% de la réaction maximale observée.
Ainsi chaque laboratoire devra fournir pour le test la définition utilisée pour
calculer le titre du sérum.
Figure 1 : Mesure du titre d’un anticorps
A – 2 – Méthodes directes de dosage des anticorps
Deux méthodes sont couramment utilisées pour mesurer la
fixation directe des anticorps sur un antigène : Le RIA (RadioImmuno
Assay), l’ELISA (Enzyme-Linked ImmunoSorbent Assay) et les techniques
d’inhibition compétitives
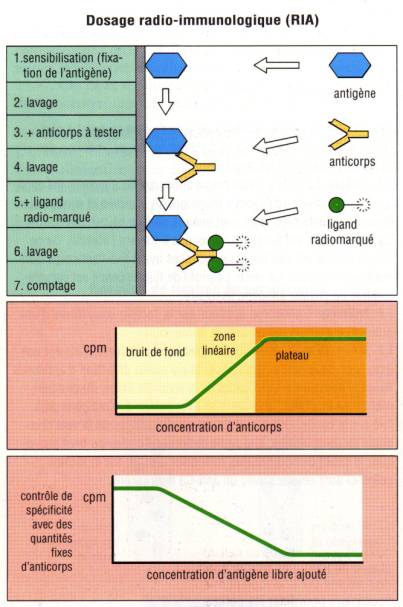
A – 2 – 1 – RIA (RadioImmuno Assay)
L’antigène spécifique des anticorps que l’on veut détecter
est préalablement marqué à l’iode 125. La préparation contenant l’anticorps est
alors incubée avec l’antigène marqué. Les complexes Ag/Ac qui se forment en
phase liquide sont alors précipités avec une solution de chlorure d’ammonium ou
de polyethylèneglycol. Le culot de précipitation est ensuite lavé avec une
solution saline et la présence de l’anticorps à doser est déterminée en
mesurant la radioactivité présente dans le précipité et en la comparant avec
une gamme étalon réalisée soit avec un sérum titré soit avec une préparation
purifiée d’anticorps de concentration connue. Cette technique bien que
relativement coûteuse et délicate car utilisant des produits radioactifs, présente
l’avantage d’une grande sensibilité. En biologie clinique, cette méthode est
encore employée pour détecter la présence d’anticorps anti-ADN natif chez les
patients atteints de LED (test de Farr) ou pour la détection des anticorps
anti-GAD chez les diabétiques.
Figure 2 : Technique de RIA
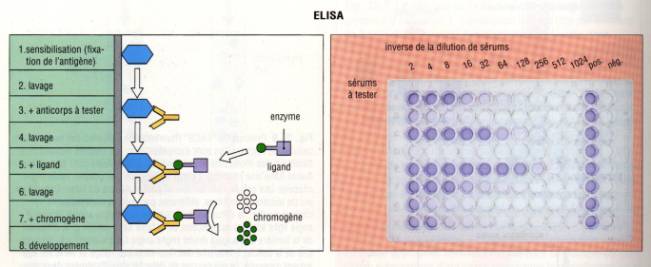
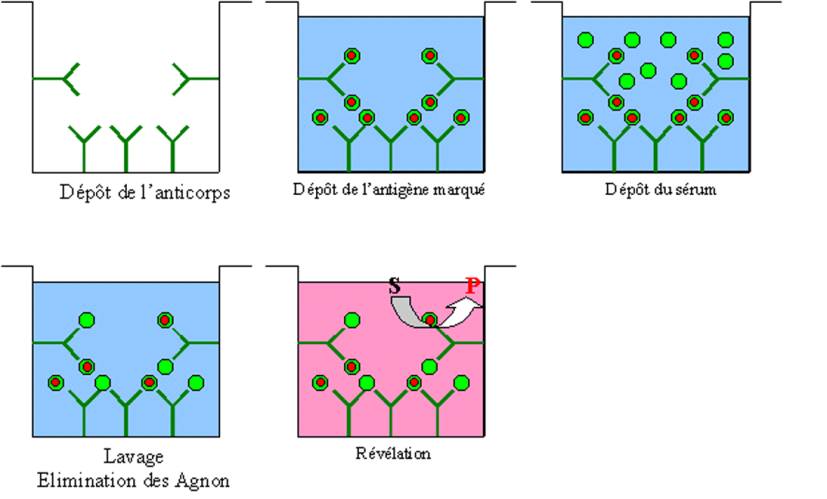
A – 2 – 2 – l’ELISA (Enzyme-Linked ImmunoSorbent Assay)
La technique ELISA, plus simple et moins coûteuse a presque
totalement remplacé le RIA. La révélation du test n’utilise pas, comme dans le
RIA de radioéléments mais est liée au clivage par une enzyme, d’un substrat
incolore en un produit coloré. Pour détecter la présence dans un sérum d’un
anticorps spécifique, l’antigène spécifique de l’anticorps à doser est déposé
dans un puits à fond plat en plastique. L’antigène est dilué dans un tampon
bicarbonate à pH 9,6 ce qui favorise les interactions électrostatiques entre
l’antigène et le plastique de la plaque et permet la fixation stable de
l’antigène au fond du puits. Des dilutions limites du sérum contenant
l’anticorps à doser sont alors déposées dans les puits. Après un temps de
contact suffisant, les puits sont lavés avec une solution saline de sorte que
seuls les anticorps spécifiques restent fixés sur l’antigène et donc au
plastique. on révèle la présence de l’anticorps fixé au fond de la plaque en
déposant ensuite dans le puit un anticorps anti-Immunoglobuline marquée avec
une enzyme qui peut être la phosphatase alcaline ou la peroxydase. Après
lavage, il ne reste plus qu’à révéler la présence des anticorps spécifiques en
ajoutant le substrat de l’enzyme ayant servie à marquer les anticorps
anti-Immunoglobuline et à lire la réaction colorée.
Figure 3 : Principes de l’ELISA
A – 2 –
3 – Techniques d’inhibition compétitives.
Dans ce type de
test, la présence et le titre d’un anticorps ou d’un antigène particulier est mesurée
par sa capacité à entrer en compétition avec un anticorps de référence marqué
par un radioélément ou une enzyme sur la fixation sur un antigène spécifique
fixé sur une plaque. La courbe étalon est déterminée en ajoutant des quantités
connues d’anticorps non marqué.
Figure 3 : Méthodes d’inhibition compétitive
A
– 3 –Dosage des anticorps par modification physique de l’antigène
La mesure directe
de la fixation d’un anticorps sur un antigène est utilisée dans la plupart des
tests sérologiques. Cependant, certains tests, important en pathologie humaine,
sont basés sur la capacité d’un anticorps de modifier les propriétés physiques
de l’antigène sur lequel il se fixe. Ces interactions secondaires peuvent être
détectées de nombreuses manières.
A
– 3 – 1 – Réaction d’agglutination.
Lorsque
l’antigène est présent à la surface d’une grosse particule, les anticorps
peuvent induire son agglutination. Elle est de réalisation particulièrement
aisée avec les bactéries et les érythrocytes, ces derniers pouvant être
utilisés directement ou comme support d’antigènes solubles fixés à leur surface
(hémagglutination passive) Ce principe est couramment utilisé pour la
détermination des groupes sanguins et est appelé réaction d’hémagglutination
directe. Dans ce cas, l’agglutination est induite en incubant des anticorps
anti-A ou anti-B avec des hématies du sujet à grouper. Si le sujet possède à la
surface de ses globules rouges l’antigène A (patient du groupe A), on observera
une agglutination avec les anticorps anti-A mais pas avec les anticorps anti-B.
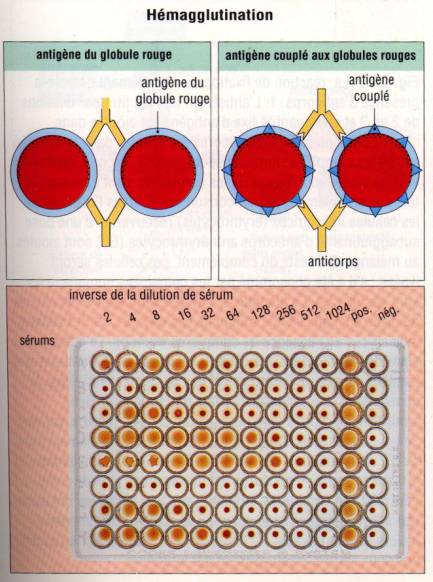
Hémagglutination passive
Des hématies ou
des particules inertes peuvent être recouvertes d’antigène et utilisées dans un
test d’hémagglutination passive. Le couplage sur des hématies peut être obtenu
par différents moyens, dont les plus classiques consistent à traiter les
hématies par l’acide tannique, ce qui les rend capables de réagir spontanément
avec des antigènes solubles comme des protéines ou des acides nucléiques, ou à
les incuber avec les antigènes en présence de glutaraldéhyde. L’inhibition de
l’hémagglutination passive par l’antigènes permet la quantification des
antigènes.
Figure 4 : Principe de l’hémagglutination
A
– 3 – 2 – Réactions de précipitation.
A
– 3 – 2 – 1 - Réaction de précipitation en milieu liquide.
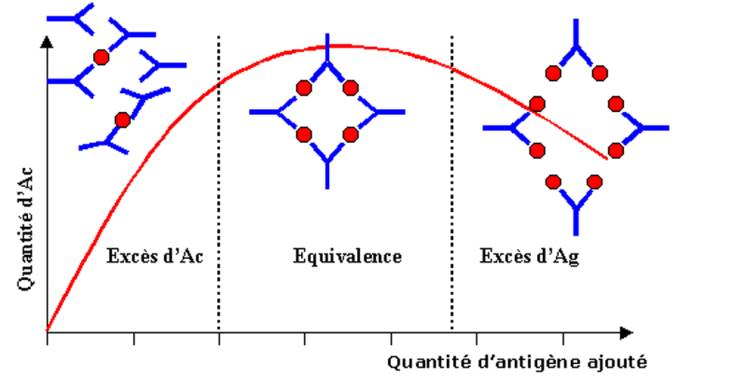
Lorsque des
quantités suffisantes d’anticorps sont mélangées avec un antigène soluble on
peut observer une réaction de précipitation. Le précipité ainsi formé est
composé de larges agrégats d’antigène reliés les uns aux autres par des
molécules d’anticorps. La quantité de précipité dépend non seulement de la
quantité d’anticorps et d’antigène mais aussi du rapport entre les deux
protagonistes. En effet, dans la réaction de précipitation, si des quantités
croissantes d’antigène soluble sont ajoutées à une quantité connue de sérum
contenant l’anticorps à doser, on observe dans un premier temps une corrélation
directe entre la quantité d’antigène apportée et la quantité de précipité. La
courbe de précipitation atteint alors un maximum et si la quantité d’antigène
augmente encore, on note que la quantité de précipité tend cette fois à
diminuer. Lorsque de faibles quantités d’antigène sont ajoutées à l’anticorps,
les complexes Ag/Ac sont formés dans des conditions ou l’anticorps est en
excès. Ainsi, chaque molécule d’Ag est couplé à plusieurs molécules
d’anticorps. Au fur et à mesure que la quantité d’antigène augmente, certains
anticorps vont pouvoir lier plusieurs antigènes différents. A la zone
d’équivalence, lorsque toutes les molécules d’Ac sont liées à deux molécules
d’antigènes différentes, il se forme alors un grand réseau qui favorise la
réaction de précipitation. Lorsque la quantité d’antigène est très élevée,
seuls de petits complexes peuvent se former, la taille réduite de ces complexes
favorise leur solubilité expliquant ainsi l’inhibition de la réaction de
précipitation observée en excès d’antigène.
Figure 5 : Précipitation Ag/Ac en milieu
liquide
La réaction de
précipitation dépend intimement du nombre de sites de fixation que chaque
anticorps possède pour l’antigène et par le nombre maximum d’anticorps qui
peuvent se fixer sur l’antigène. Ces quantités sont définies comme étant la
valence de l’anticorps ou de l’antigène. La valence de l’antigène ou de
l’anticorps doit être au moins égale à deux pour qu’une réaction de
précipitation puisse avoir lieu. La valence d’un anticorps dépend de sa classe
et varie de 2 (IgA,G,E et D) à 10 (IgM). L’antigène ne sera précipité que s’il
possède au moins deux sites de fixation pour l’anticorps. Cette condition est généralement
obtenue avec des antigènes macromoléculaires dont la structure complexe permet
la fixation de nombreux anticorps de spécificités diverses. Le site de
l’antigène sur lequel vient se fixer un anticorps est appelé déterminant
antigénique ou épitope.
Néphélémétrie
La
précipitation de complexes antigène/anticorps en milieu liquide peut permettre
un dosage très précis des antigènes. En effet, la formation de ces complexes
entraîne une augmentation de la turbidité du milieu qui, à concentration
d’anticorps constant, ne dépend que de la quantité de l’antigène à doser. Les
variations de turbidité du milieu sont mesurées à l’aide d’un néphélémètre. Cet
appareil possède un rayon laser dont le faisceau traverse la cuve où à lieu la
réaction antigène/anticorps. L’augmentation de turbidité du milieu entraîne une
déviation du faisceau qui est analysée par des photomultiplicateurs et comparée
à la déviation obtenue avec des quantités connues d’antigène. On en déduit la
quantité d’antigène présente dans l’échantillon à doser. Ce principe est
couramment employé pour doser une multitude de protéines sériques comme par
exemples les immunoglobulines ou les fractions du complément.
A
– 3 – 2 – 2 - Réaction de précipitation en milieu gélifié.
Lorsqu’un
antigène et un anticorps sont introduits dans un milieu gélifié en des points
différents, ils diffusent et des précipités peuvent se former au point de
rencontre si le rapport des concentrations d’antigène et d’anticorps s’y prête.
On distingue, selon le type de support sur lequel le gel est appliqué,
l’immunodiffusion en tubes ou en plaques.
Immunodiffusion en tube
C’est
historiquement la première des techniques d’immunoprécipitation en gel
puisqu’elle a été décrite par Oudin en 1946. Le principe de la technique consiste
à remplir un tube de verre avec un gel d’agar dans lequel un anticorps a été
préalablement incorporé, puis à appliquer une solution d’antigène dans le tube.
L’antigène progresse rapidement par simple diffusion dans le gel en créant un
gradient de concentration. Si la concentration initiale d’antigène est
suffisante, un précipité se forme au niveau du front de progression de
l’antigène.
Immunodiffusion
sur plaque (technique d’Ouchterlony)
L’adaptation en
plaque de verre de la technique d’immunodiffusion en tube a représenté un
progrès technique important. Les tubes sont remplacés par des plaques dans
lesquelles sont creusés des puits. Les solutions d’antigènes et d’anticorps,
placées dans des puits différents, diffusent librement dans le gel et donnent
lieu à des précipités à la zone d’équivalence. Cette technique en plaque est
plus simple à mettre en œuvre que la technique en tube. Elle a surtout
l’avantage de permettre la comparaison directe de différentes préparations
d’antigène. Il suffit pour cela de placer les diverses préparations
antigéniques dans différents puits disposés sur un cercle dont le centre est
creusé d’un puits où la solution d’anticorps est introduite.
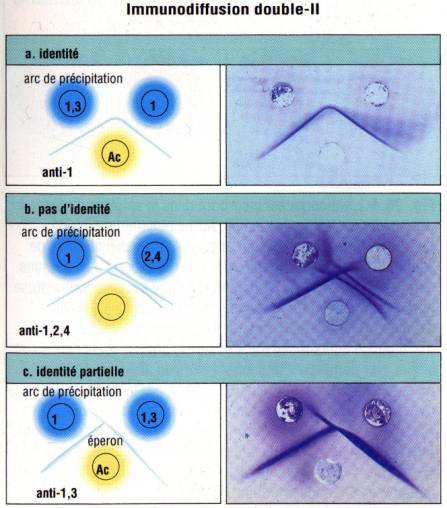
Lorsqu’une
préparation d’antigène est introduite simultanément dans deux puits adjacents,
les traits de précipitation vont se rejoindre et fusionner. C’est la réaction
d’identité. Si au contraire, deux solutions antigéniques différentes sont
disposées dans deux puits adjacents, les traits de précipitation vont se
couper. C’est la réaction de non identité. Enfin, si deux solutions
antigéniques donnent lieu à des réactions croisées, les bandes vont fusionner
mais on note cependant au delà du point de fusion des deux précipités, une
projection qui prolonge le précipité formé par l’antigène. C’est la réaction
d’identité partielle.
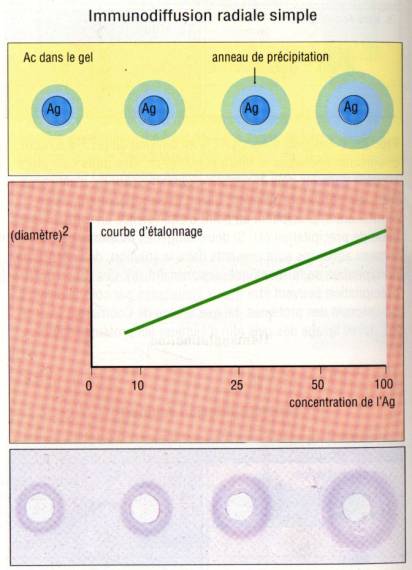
Immunodiffusion
radiale (Technique de Mancini)
Un gel d’agar
dans lequel un anticorps a été préalablement incorporé est déposé sur une lame
de verre. La solution d’antigène qui est déposée dans un puits diffuse dans
l’agar et donne lieu à la formation d’un halo de précipitation dont le diamètre
extérieur est proportionnel à la concentration initiale d’antigène. Cette
technique a été proposée par Mancini pour doser certains antigènes. Il suffit
en effet de calibrer la méthode en utilisant des quantités connues d’antigène
purifié.
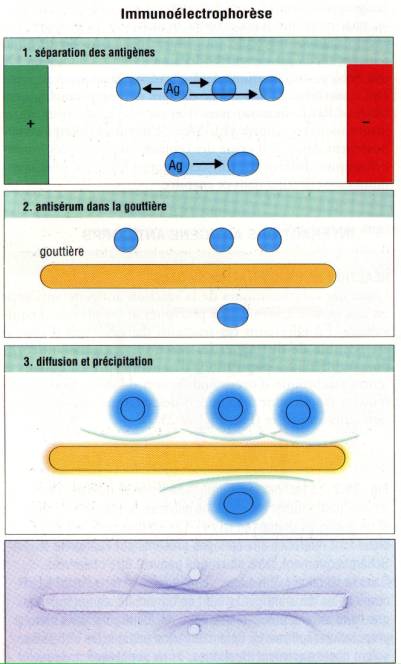
Immunoélectrophorèse
(Grabar et Williams)
Une autre
application simple et très discriminative de l’immunodiffusion est l’immunoélectrophorèse.
Le principe de la technique consiste à introduire un mélange d’antigènes dans
un puits creusé dans une plaque d’agar et à appliquer un champ électrique
pendant une à deux heures afin de séparer les molécules d’antigène selon leur
mobilité électrophorétique. Le champ électrique est alors coupé et un antisérum
polyspécifique est introduit dans une rigole parallèle au champ de migration de
la préparation d’antigène. Les anticorps et les antigènes diffusent alors
librement les uns vers les autres et donnent lieu à des précipités analogues à
ceux décrits dans la méthode d’Ouchterlony. L’immunoélectrophorèse s’est
révélée particulièrement spectaculaire pour l’analyse des protéines du sérum,
permettant d’objectiver plus de 30 molécules différentes. De plus cette
technique à longtemps été utilisée pour détecter la présence d’une
immunoglobuline monoclonale dans le sérum de patient atteint par exemple de
myélome multiple.
Immunoélectrophorèse
en fusée (Technique de Laurell)
Dans cette
technique, un anticorps spécifique de l’antigène à doser est incorporé dans une
plaque de gel. Différentes dilutions de la préparation antigénique à doser sont
réparties dans des puits alignés. Un courant électrique est alors appliqué
perpendiculairement à la ligne des puits. Un halo en forme en fusée se forme et
progresse tant que l’antigène est en excès. L’existance d’une relation linéaire
entre la concentration d’antigène dans les puits et la hauteur du précipité
permet d’utiliser cette technique pour un dosage très précis et très sensible
des antigènes.
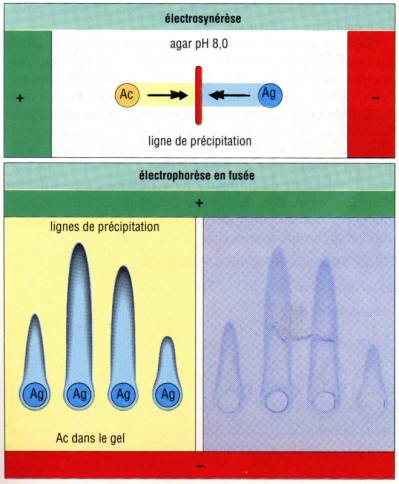
Contre-Immunoélectrophorèse
(ou électrosynérèse)
C’est une
technique proche de la précédente, où l’anticorps est distribué dans une ligne
de puits parallèle à celle des puits contenant les antigènes. Un courant est
appliqué perpendiculairement aux lignes des puits. Lorsque l’anticorps et
l’antigène se rencontrent, il se forme un arc de précipitation.
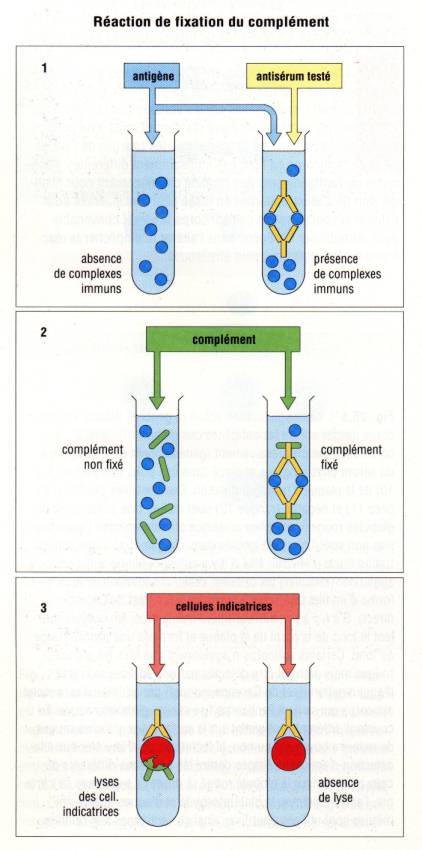
A – 4 – Réactions utilisants le complément
De nombreuses
réactions sérologiques utilisent l’aptitude du C1q puis des autres composés du
complément à se fixer sur les complexes immuns. Le complément présent dans le
sérum initial est inactivé par chauffage à 56°C pendant 30 minutes, ce qui
détruit le C1. Du complément est obtenu à partir d’une source standardisée. La
fixation de ce complément peut être quantifiée en dosant la quantité de
complément consommée au cours de la technique de déviation du complément. Au
cours de cette méthode, une préparation antigénique étalonnée est incubée avec
le sérum à tester et préalablement inactivé par chauffage. Du complément titré
par dosage hémolytique est alors ajouté à la réaction et l’ensemble de ces
réactifs est porté à 37°C pendant 30 minutes. A la fin de cette phase dite de
fixation, on ajoute des hématies de mouton et un sérum anti-érythrocyte de
mouton étalonné. Après une courte période d’incubation, on dose le complément
consommé par la réaction. Si le sérum contient des anticorps fixant le
complément et dirigés contre la préparation antigénique, l’activité du
complément sera abaissée et l’hémolyse des hématies de mouton sera diminuée
proportionnellement à cet abaissement.
B – Utilisation d’anticorps anti-Immunoglobuline
Comme nous
l’avons vu précédemment, un anticorps peut être détecté par fixation directe de
cet anticorps préalablement marqué sur son antigène spécifique fixé sur un
plaque de plastique. Une approche plus générale qui permet d’éviter le marquage
préalable de chaque préparation d’anticorps consiste à détecter l’anticorps
spécifique non marqué fixé sur son antigène immobilisé en utilisant des
anticorps anti-Immunoglobuline (Ac anti-Ig) marqués. Des Ac anti-Ig peuvent
être obtenus en immunisant des animaux avec des préparations pures
d’immunoglobulines. Les Ac anti-Ig produits par l’animal sont alors purifiés du
sérum par chromatographie d’affinité. Ces anticorps sont alors marqués avec un
radioélément ou avec une enzyme puis utilisés comme réactifs pour détecter des
anticorps fixés.
Robin Coombs a
été le premier à utiliser des Ac anti-Ig pour détecter les anticorps
responsables de la maladie hémolytique du nouveau né. Les femmes Rhésus négatif
peuvent s’immuniser contre cet antigène (Rhésus) si elles sont exposées, le
plus souvent au moment de l’accouchement, avec des hématies fœtales Rhésus+.
Dans ce cas, la femme Rhésus- va produire des anticorps anti-Rhésus. Si au
cours d’une grossesse ultérieure, le fœtus est Rhésus+, les IgG anti-Rhésus de
la mère vont pouvoir traverser la barrière placentaire, se fixer sur les
hématies du fœtus qui seront alors détruites par les cellules phagocytaires du
foie fœtal induisant par la même une anémie hémolytique chez le fœtus ou le
nouveau-né. La densité des antigènes Rhésus à la surface des hématies étant
faible, les anticorps anti-Rhésus ne peuvent induire l’agglutination des
hématies. Ainsi, la détection directe de ces anticorps a été impossible jusqu’à
l’utilisation des Ac anti-Ig. Les IgG maternelles fixées sur les globules
rouges du fœtus peuvent en effet être détectées si, après lavage des hématies
fœtales, on ajoute des Ac anti-Ig. Ceux-ci vont induire l’agglutination des
hématies fœtales sur lesquelles sont fixés les anticorps maternels. Ce test est
appelé test de Coombs direct car il permet la détection directe d’anticorps
fixés sur les cellules fœtales. Il existe aussi un test de Coombs indirect qui
permet de détecter chez la mère la présence d’Ac anti-Rhésus. Dans ce test, le
sérum de la mère est d’abord incubé avec des hématies Rhésus+, si la mère
possède des anticorps anti-Rhésus, ceux-ci vont se combiner aux hématies. Après
lavage des globules rouges, on ajoute les Ac anti-Ig qui induisent alors
l’agglutination des hématies.
Les Ac anti-Ig
sont largement utilisés pour le diagnostic et la recherche. Des Ac anti-Ig
marqués peuvent être utilisés dans les techniques de RIA ou d’ELISA pour
détecter des anticorps fixés sur des antigènes immobilisés. Des Ac anti-Ig
spécifiques de chaque isotype d’immunoglobuline peuvent être produits en
immunisant des animaux avec des préparations pures de chaque isotype. Ces
anticorps peuvent ensuite être utilisés pour mesurer combien un anticorps
d’isotype particulier peut se fixer sur un antigène donné et ainsi rendre
compte du caractère approprié ou non de la réponse immunitaire. De plus, la
détermination de l’isotype de l’anticorps spécifique de l’antigène revêt une
importance toute particulière en allergologie dans la détermination de la
présence d’IgE spécifique d’un allergène donné.
L’utilisation
des Ac anti-Ig spécifique de chaque isotype d’immunoglobuline a permis de faire
des progrès considérables dans la détection d’immunoglobulines monoclonales
dans le sérum des patients rendant du même coup quasi obsolète la précédente
technique d’immunoéléctrophorèse dans cette application. Cette technique a en
effet été remplacée par la méthode dite d’immunofixation. Dans celle-ci, les
protéines du sérum sont soumises à une électrophorèse en gel d’agarose puis
incubées directement avec un anticorps anti-Ig totales, anti-IgG, anti-IgA et
anti-IgM qui, après révélation au bleu de Coomasie, indiquera la présence de
ces différentes immunoglobulines. Si le sérum du patient contient une
concentration importante d’Ig monoclonales, l’homogénéité structurale de cette
protéine va faire que toutes les molécules d’Ig monoclonales vont migrer sur le
gel au même endroit. De plus, toutes les molécules d’Ig monoclonales ont le
même isotype ce qui implique que seuls les Ac anti-Ig de l’isotype de l’Ig
monoclonale vont pouvoir se fixer sur cette protéine. Enfin, la concentration
importante de cette Ig monoclonale dans le sérum du patient va faire qu’elle
deviendra détectable après coloration du gel au bleu de Coomasie et apparaîtra
sur le gel comme un trait fin coloré en bleu dans la piste où à été déposé l’Ac
anti-Ig dirigé contre l’isotype de la protéine monoclonale. Cette distinction
isotypique des Ig monoclonales a une très grande importance en pathologie
humaine. En effet, certaines maladies comme le myelome multiple sont associées
à la présence dans le sérum des patients d’Ig monoclonale d’isotype IgG ou IgA
alors que les patients atteint de maladie de Waldenström ont dans le sérum des
Ig monoclonale d’isotype IgM.
C – Les anticorps monoclonaux
Les anticorps
générés au cours d’une réponse immune naturelle ou après immunisation sont en
fait un mélange de molécules de spécificité et d’affinité différentes. Une part
de cette hétérogénéité résulte de la production d’anticorps reconnaissant des
épitopes différents à la surface de l’antigène. Mais, même lorsque les Ac
reconnaissent le même épitope, il existe encore une certaine hétérogénéité qui
peut d’ailleurs être mise en évidence par des différences de point
isoélectrique entre les différents anticorps produits. Les Ac anti-Ig
polyclonaux sont d’une grande utilité en biologie humaine mais ils présentent un
certain nombre d’inconvénients liés à l’hétérogénéité des anticorps qu’ils
contiennent. D’une part, chaque lot d’Ac anti-Ig est différent et ce même si
une préparation antigénique identique a été utilisée pour immuniser selon un
protocole standardisé un animal génétiquement identique. De plus, les volumes
de réactif obtenus par cette méthode sont limités rendant impossible
l’utilisation d’un même lot d’ Ac anti-Ig lorsque l’on souhaite réaliser des
tests sérologiques au long cours. Enfin, certains antisérums peuvent contenir
des anticorps ayant une réactivité croisée avec d’autres antigènes que celui
ayant servi pour immuniser l’animal. Pour toutes ces raisons, il a été
nécessaire de mettre au point une méthode permettant d’obtenir en quantité
illimitée des anticorps de structure homogène et de spécificité donnée.
Il était connu
de longue date que chez les patients atteints de myélome multiple on observe un
envahissement médullaire de plasmocytes tumoraux. Les plasmocytes correspondent
à l’étape de différenciation ultime des lymphocytes B. C’est au stade
plasmocyte que les cellules B produisent des anticorps. Ainsi, chez les malades
atteints de myélome multiple, la prolifération plasmocytaire se traduit par la
présence dans le sérum des patient d’un pic à l’électrophorèse des protéines au
niveau des gammaglobulines. Les plasmocytes tumoraux derivant tous d’une même
cellules l’anticorps produit dans le sérum des patient est clonal et on parle
donc de pic d’immunoglobuline monoclonal. Ces cellules myélomateuses de par
leurs propriétés prolifératives et de synthèse d’anticorps représentaient donc
une cellule de choix pour produire des anticorps monoclonaux si tant est que
l’on était capable de leur faire produire l’immunoglobuline souhaitée. C’est à
ce niveau que réside toute l’ingéniosité de la découverte de Köhler et
Milstein. Ces chercheurs ont en effet mis au point une technique permettant de
produire une population homogène d’anticorps de spécificité donnée en
produisant un hybride entre un lymphocyte B immun et une cellule tumorale.
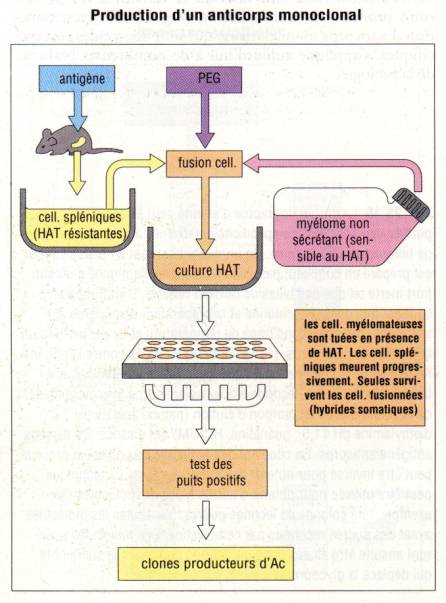
De façon
pratique, Ils ont réalisé cet exploit en fusionnant des cellules spléniques de
souris immunisée avec des cellules myélomateuses de souris. Les cellules
spléniques apportent la capacité de produire un anticorps spécifique alors que
les cellules myélomateuses outre leur caractère tumorale qui assure la
prolifération quasi infinie de l’hybride apporte aussi toute la machinerie
cellulaire nécessaire à la production de l’immunoglobuline monoclonale. En
outre, l’utilisation de cellules myélomateuses ne produisant pas par elle-même
d’immunoglobulines fait que l’anticorps produit par l’hybride vient seulement
de la cellule splénique. La fusion des deux cellules se fait en utilisant du
polyethylèneglycol. Après la fusion, les cellules hybrides sont sélectionnées
en utilisant une drogue qui tue les cellules myélomateuses n’ayant pas
fusionné. De leur coté, les cellules spléniques non fusionnées ont une demi-vie
très courte et meurent naturellement quelques jours après la fusion. Finalement,
seuls les hybrides survivent. Les hybridomes ainsi formés produisant un
anticorps de spécificité voulue sont alors identifiés et cloné. Ainsi, chaque
hybridome est un clone qui dérive de la fusion d’une seule et unique cellule B.
De ce fait, les anticorps produits par cet hybridome sont tous identiques tant
en terme de spécificité que de structure. Ces anticorps sont appelés anticorps
monoclonaux. Cette technologie a révolutionné l’utilisation des anticorps en
médecine et sont actuellement largement utilisés dans la plupart des tests
sérologiques et même comme agent thérapeutique.
D – Mesure de l’affinité des anticorps
L’affinité d’un
anticorps représente la force de fixation entre un ligand monovalent et un
épitope unique.
L’affinité d’un anticorps
peut être déterminée directement par la technique de l’équilibre de dialyse.
Une quantité connue d’anticorps est placée dans un boudin de dialyse dont la
taille des pores ne permet pas la diffusion de l’anticorps dans le bain de
dialyse. Des molécules d’antigène spécifique de l’anticorps sont placées dans
le bain de dialyse. La petite taille de l’antigène lui permet de diffuser
librement à travers la membrane et donc de se fixer sur l’anticorps. Les
molécules d’antigène complexées à l’anticorps ne peuvent plus diffuser
librement et restent dans le boudin de dialyse. La mesure de la concentration
en antigène dans le boudin et dans le bain de dialyse permet de déterminer la
quantité d’antigène fixé sur l’anticorps et ainsi de déterminer l’affinité de
l’antigène pour l’anticorps. Les résultats sont généralement analysés en
effectuant une courbe de Scatchard.
Alors que
l’affinité mesure la force de liaison entre un déterminant antigénique et un
site de fixation pour l’antigène, dans le cas d’un antigène possédant de
multiple site antigéniques identiques l’anticorps peut interagir avec
l’antigène par l’intermédiaire de l’ensemble de ses sites de fixation pour
l’antigène. Ces multiples interactions possibles entre un anticorps et un
antigène augmentent considérablement les forces de liaisons. La force globale
de fixation d’un anticorps pour un antigène est appelé avidité. Ainsi, pour les
IgM, chaque site de fixation pour l’antigène a une affinité faible, mais comme
les IgM possèdent 10 sites de fixation l’avidité des IgM pour leur antigène est
relativement forte.
Bien que la
mesure de l’affinité des anticorps soit réservée au domaine de la recherche, la
mesure de l’avidité d’anticorps spécifique d’antigènes infectieux est très
utilisée en clinique humaine. La méthode employée pour réaliser cette mesure
est une adaptation de la technique ELISA. L’antigène du micro-organisme est
fixé au fond d’un puits en plastique. Le sérum du patient est alors ajouté. Si
celui-ci contient des anticorps spécifiques du pathogène, ceux-ci vont se fixer
sur l’antigène accroché au plastique. Dans la technique ELISA classique la
présence de l’anticorps est révélée par un Ac anti-Ig marqué par une enzyme et
convertion d’un substrat spécifique. Pour la mesure de l’avidité, on ajoute une
étape supplémentaire. Après avoir ajouté le sérum du patient, les puits sont
lavés avec un agent chaotropique (généralement de l’urée fortement concentrée)
ce qui a pour conséquence de décrocher tous les anticorps de faible avidité. Ce
test ne révèle donc que les Ac spécifiques de forte affinité. La comparaison de
ce résultat avec celui obtenu avec la technique ELISA standard permet de savoir
si le sérum du patient contient des anticorps de forte affinité spécifiques du
micro-organisme. Cette détermination est particulièrement importante pendant la
grossesse. En effet, durant cette période, la femme peut contracter des
infections susceptibles d’être transmises au fœtus et causant pour certaines
d’entre elles des malformations voir des morts in utéro. Il est donc primordial de surveiller le statut
sérologiques des femmes enceintes vis à vis de ces micro-organismes.
Généralement, la présence d’IgM dans le sérum de la mère signe une infection en
cours alors que la présence d’IgG signe plutôt une infection ancienne.
Cependant, la présence transitoire des IgM fait que chez certaine femme ayant
une infection active on ne détecte que des IgG ce qui rend impossible le
diagnostic d’infection récente ou ancienne. Un des moyens pour répondre à cette
question consiste à mesurer l’avidité des anticorps produits. Dans le cas d’une
infection active récente l’avidité des IgG sera faible alors que dans le cas
d’une infection ancienne l’avidité sera forte.
E – Utilisation des anticorps dans la détection d’antigènes cellulaires ou tissulaires.
Les anticorps
ayant la propriété de ce fixer de façon stable et spécifique sur un antigène,
ces molécules peuvent être utilisés pour détecter la présence d’un antigène
particulier sur une cellule ou dans un tissu. Tout comme dans les tests
sérologiques, l’anticorps se fixe sur son antigène cible de façon stable ce qui
permet par simple lavage d’éliminer les anticorps non fixés et de révéler
uniquement la fixation spécifique de l’antigène. La plupart des anticorps
reconnaissent des protéines antigèniques sous forme native (épitope
conformationnel) ce qui nécessite que la structure tridimensionnelle de
l’antigène soit préservée si l’on veut que celui-ci soit reconnu par
l’anticorps. Dans ce cas, on utilisera un substrat antigénique : cellules
ou coupes de tissu non fixées. Certains anticorps se combinent au contraire
avec l’antigène dénaturé (épitope linéaire). Ces anticorps devront être
utilisés sur des coupes de tissu ou des cellules fixées. L’anticorps une fois
fixé sur l’antigène cellulaire ou tissulaire peut être visualisé en utilisant
des méthodes de révélation extrêmement variées.
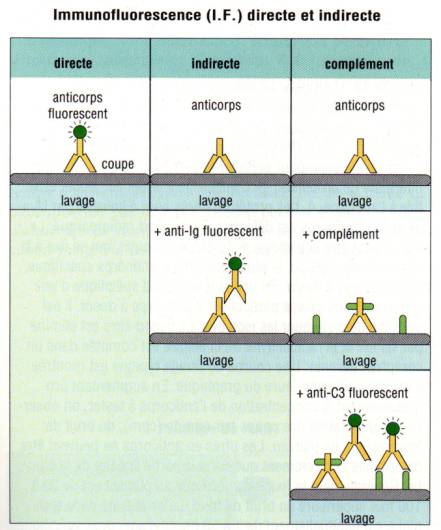
E – 1 – Immunofluorescence directe et indirecte
Une des
technique de détection les plus performantes pour détecter la présence d’un
anticorps fixé sur un antigène tissulaire ou cellulaire est
l’immunofluorescence. Dans cette technique, un colorant fluorescent
(l’isothiocyanate de fluorescéine ou la phycoérythrine) est fixé de façon
covalente à l’anticorps spécifique et permet la détection directe de l’antigène
à analyser. On peut en outre utiliser des Ac anti-Ig fluorescents pour détecter
des anticorps fixés sur l’antigène tissulaire on parle alors dans ce cas
d’immunofluorescence indirecte. La lecture des coupes tissulaires ou des
cellules ainsi marquées est réalisée à l’aide d’un microscope UV à
fluorescence.
E – 2 – Immunohistochimie
L’immunohistochimie
est une méthode alternative à l’immunofluorescence pour détecter la présence
d’un antigène sur une coupe de tissu. Dans ce cas, l’anticorps de révélation
n’est pas couplé à un fluorochrome mais à une enzyme qui convertie un substrat
incolore en un produit coloré insoluble qui se dépose au niveau de l’anticorps.
Le résultat peut être observé directement en microscopie optique.
E – 3 – Méthodes électrophorétiques
E
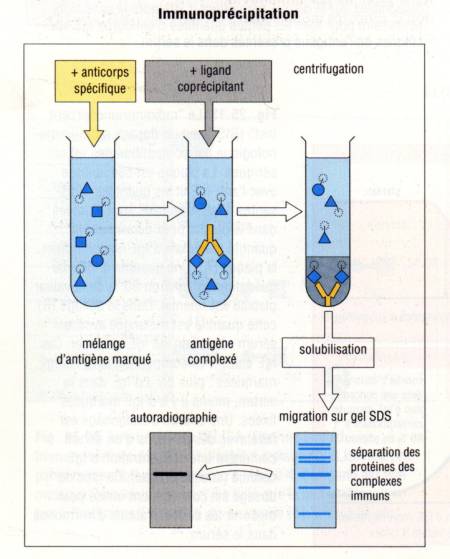
– 3 – 1 – Immunoprécipitation
Si ces
techniques permettent d’observer directement ou indirectement si un anticorps
se fixe sur un antigène cellulaire ou tissulaire, on peut aussi utiliser les
anticorps pour déterminer exactement la nature de l’antigène reconnu par un
anticorps. Plusieurs méthodes permettent d’atteindre ce but. La première
d’entre elles est la technique d’immunoprecipitation. Dans cette technique, les
cellules contenant l’antigène à analyser sont incubées en présence d’acide
aminé marqué. Ainsi, l’ensemble des protéines synthétisées par la cellule
seront radioactives. Les cellules ainsi marquées sont lysées à l’aide d’un
détergeant. L’anticorps spécifique de l’antigène à analyser fixé sur des billes
d’agarose est alors ajouté au lysat cellulaire. Le complexe Ag/Ac est alors
précipité, le culot de précipitation lavé pour éliminer les Ag non fixés.
L’antigène est alors élué de l’anticorps par un détergent ionique : le
dodecysulfate de sodium (SDS). Non seulement le SDS dissocie l’Ag de l’Ac mais
il charge aussi négativement l’antigène. La préparation antigénique ainsi
obtenue est alors déposée au sommet d’un gel de polyacrylamide pour y subir une
électrophorèse (PAGE : PolyAcrylamide Gel Electrophoresis). Après
application d’un courant électrique, les protéines antigéniques à analyser
migrent vers le pole +. Le SDS chargeant de façon homogène l’ensemble des
protéines, leur migration ne dépendra plus de leur charge mais uniquement de
leur masse moléculaire. La position des protéines sur le gel, direct reflet de
leur masse moléculaire est révélée par autoradiographie.
Cette technique
de SDS-PAGE peut être combinée à une isoélectrofocalisation dans une méthode
appelée électrophorèse bidirectionnelle. Dans ce cas, les protéines
immunoprécipitées sont éluées dans un tampon non ionique à base d’urée et
déposées sur un gel d’IEF qui permet la séparation des protéines en fonction de
leur point isoélectrique. Apres la migration, le gel est démoulé et placé au
sommet d’un gel de polyacrylamide contenant du SDS. Au cours de cette seconde
étape, les protéines se séparent en fonction de leur masse moléculaire. Cette
méthode augmente considérablement la sensibilité de détection et le pouvoir de
résolution du SDS-PAGE.
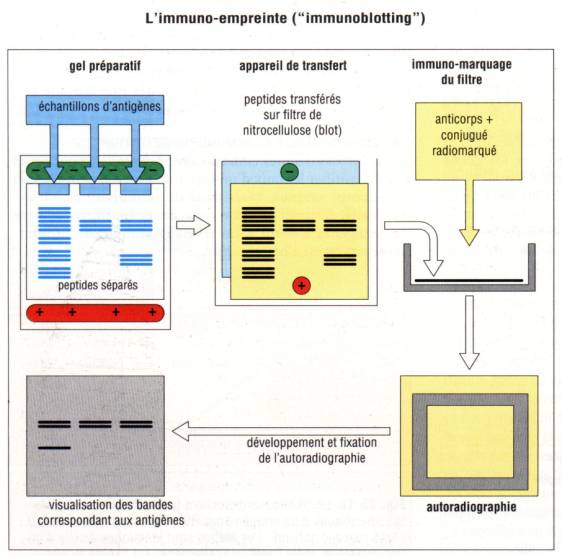
E – 3 – 1 – Immunoempreinte ou Western
blot
Une approche
alternative pour éviter les problèmes liés à la manipulation de substances
radioactives consiste à lyser les cellules directement dans un tampon riche en
détergent. Le lysat cellulaire ou tissulaire ainsi obtenu est incubé avec du
SDS et soumis à une électrophorèse en gel de polyacrylamide. Les protéines
ainsi séparées en fonction de leur masse moléculaire sont alors transférées sur
une membrane de nitrocellulose. Les membranes sont ensuite incubées avec un
anticorps marqué spécifique. Après lavage pour éliminer la fixation d’anticorps
non spécifique, la position des protéines est révélée à l’aide d’un Ac anti-Ig
marqué avec un radioélément ou une enzyme. Cette méthode appelée Western blot a
de nombreuses applications en biologie clinique. Elle permet notamment de
détecter la présence dans le sérum des patients HIV+ d’anticorps spécifiques du
virus et donc de contribuer au diagnostic de l’infection.
III – Etude de la réponse à médiation cellulaire
A – Définitions
L’analyse de la
réponse immunitaire repose en pratique clinique quotidienne sur l’étude de la
réponse humorale et ce essentiellement pour des raisons pratiques. Les
anticorps sont en effet les produits les plus accessibles de la réponse immunitaire
adaptative. Cependant, les lymphocytes T interviennent à toutes les étapes, ce
qui rend l’étude du devenir de cette population cellulaire indispensable.
L’étude des lymphocytes nécessite au préalable leur séparation des autres
éléments figurés du sang. Enfin, les différentes sous-populations
lymphocytaires identifiées sur leurs différentes propriétés fonctionnelles
doivent être analysées.
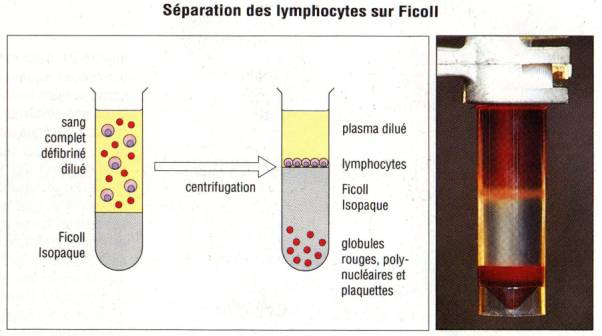
B – Isolement des lymphocytes
Les lymphocytes
peuvent être isolés du sang périphérique, de la moelle osseuse hématopoïétique,
des organes lymphoïdes, des épithélium et des sites inflammatoires. La première
étape consiste à séparer les lymphocytes des autres populations leucocytaires
et des globules rouges par centrifugation sur gradient de densité. A cet effet,
des solutions sont disponibles dans le commerce comme le métrizamide ou le
Ficoll. La centrifugation du sang total sur ces solutions permet de séparer un
anneau à l’interface entre le gradient et le plasma contenant des lymphocytes
et des monocytes. Bien que les lymphocytes du sang périphérique soient les plus
accessibles, ils ne sont pas forcement représentatifs du système lymphoïde
puisque seuls les lymphocytes recirculant peuvent être isolés du sang. Chez
l’animal, les lymphocytes sont plutôt isolés des organes lymphoïdes secondaires
comme la rate ou les ganglions mais aussi des organes lymphoïdes primaires
comme la moelle osseuse hématopoïétique ou le thymus, voire des épithélia.
Enfin, les lymphocytes peuvent être collectés directement par ponction au niveau
d’un site inflammatoire c’est le cas du liquide synoviale dans la polyarthrite
rhumatoïde.
C – Etude
phénotypique des sous-populations lymphocytaires
C – 1 – Généralités
Les lymphocytes
au repos présentent une apparence uniforme. Ce sont des cellules rondes à noyau
volumineux et dense et à cytoplasme peu abondant. Malgré ces caractéristiques
morphologiques communes, les lymphocytes peuvent être divisés en
sous-populations cellulaires aux caractéristiques fonctionnelles très
différentes. On peut en effet différencier ces sous populations lymphocytaires
en étudiant l’expression de protéines membranaires propres à chaque type de
lymphocyte. Ainsi, les lymphocytes B et T peuvent être distingués par des
anticorps reconnaissant des régions conservées des récepteurs pour l’antigène
présents sur ces deux types cellulaires. Les lymphocytes T peuvent eux-mêmes
être subdivisés en lymphocytes T auxiliaires et cytotoxiques sur la base de
leur expression membranaire en protéine CD4 ou CD8.
C – 2 – Analyse des sous populations lymphocytaires par cytométrie en flux
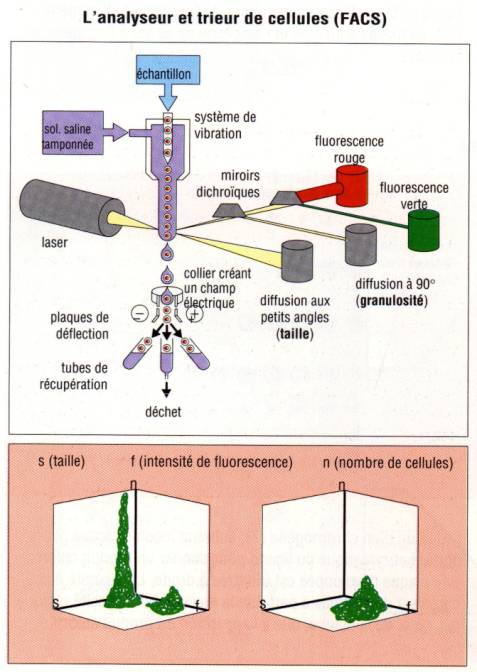
La cytométrie
en flux a permis de faire des progrès considérables dans l’étude des
différentes sous-populations lymphocytaires. Cet appareil est utilisé pour
identifier, voire pour étudier les propriétés fonctionnelles, de sous
populations cellulaires à l’intérieur d’une population cellulaire hétérogène.
L’identification des cellules repose sur l’utilisation d’anticorps monoclonaux
marqués par un fluorochrome et spécifiques d’une protéine membranaire propre à
la cellule à analyser. La première étape de la réaction consiste donc à incuber
une population cellulaire avec un anticorps marqué qui se fixe spécifiquement
sur une sous population cellulaire donnée. Après lavage dans une solution
saline, les cellules sont analysées par cytométrie en flux. Dans cet appareil,
les cellules sont aspirées dans une gaine liquide. Cette gaine liquide se
termine par un fin capillaire ne permettant le passage que d’une cellule à la
fois et donc d’analyser les cellules une par une. La cellule ainsi isolée passe
devant le faisceau d’un rayon laser. Le passage de la cellule devant le
faisceau laser à plusieurs conséquences. La première est d’induire une
déviation du faisceau et la seconde est d’exciter les fluorochromes fixés aux
anticorps et donc aux cellules à détecter et ainsi d’induire l’émission d’une
fluorescence. Des photomultiplicateurs détectent les déviations du faisceau
laser qui renseigne à la fois sur la taille et la granulosité des cellules. Ils
détectent aussi les émissions de fluorescence indiquant la présence de
l’anticorps sur la cellule et renseignent donc sur l’expression des divers
molécules de surface présentes sur les sous populations lymphocytaires.
Certains cytomètres sont également équipés d’un trieur de cellule. Ce
dispositif permet de séparer et d’isoler la sous population lymphocytaire afin
de faciliter son étude ultérieure. Lorsque les cellules sont marquées avec un
seul anticorps fluorescent, les résultats du cytomètre sont généralement
exprimés sous forme d’histogramme représentant en abscisse l’intensité de
fluorescence et en ordonné le nombre de cellules analysées. Si au moins deux
anticorps sont employés pour marquer les cellules (chacun des fluorochromes
ayant des spectres d’émission différents), les résultats sont alors exprimés
sous la forme d’un nuage de point avec en abscisse l’intensité de fluorescence
du premier fluorochrome et en ordonné l’intensité de fluorescence du second.
L’analyse par cytométrie en flux est couramment utilisée en immunologie. Par
exemple, cet appareil a joué un rôle dans l’identification des sous populations
lymphocytaires atteintes au cours de l’infection par le virus HIV.
C – 3 – Méthodes physiques de séparation et d’analyse des sous populations lymphocytaires
Bien que le
cytomètre en flux soit particulièrement bien adapté à l’isolement d’un nombre
restreint de cellules, lorsque l’on désire séparer rapidement une grande
quantité de cellules les méthodes de séparation mécanique sont bien plus
adaptées. Une méthode simple et élégante pour séparer des populations
cellulaires consiste à utiliser des anticorps marqués par des billes
magnétiques. Après incubation des cellules avec l’anticorps celui-ci se fixe
spécifiquement sur une protéine membranaire présente à la surface des cellules
à séparer. Les cellules sont alors déposées sur une colonne sur laquelle est
appliqué un puissant champs électrique. A ce niveau, les cellules ayant fixé
l’anticorps voient leur migration interrompue alors que les cellules non
marquées migrent au travers de la colonne et sont éliminées. La colonne est
alors lavée plusieurs fois avec une solution saline pour éliminer toute les
cellules non fixées puis le champ magnétique est interrompu et les cellules qui
étaient fixées migrent sur la colonne et sont récupérées.
Des méthodes
encore plus simples peuvent être utilisées cependant la pureté de la population
cellulaire à séparer est moins bonne. Ainsi, la technique du panning consiste à
fixer sur une boîte de Pétri un anticorps spécifique d’une molécule de surface
présente sur la cellule à séparer. La suspension cellulaire est déposée dans la
boîte et incubée quelques heures à 37°C. Les cellules à séparer se fixent sur
l’anticorps et se retrouvent donc accrochées au fond de la boîte de Pétri.
Après plusieurs lavages en solution saline pour éliminer les cellules non
fixées, il ne reste plus qu’à récupérer les cellules adhérentes. Une technique
alternative consiste à éliminer les cellules indésirables en incubant la
suspension cellulaire avec des anticorps monoclonaux spécifique de la molécule
de surface présente sur ces cellules et d’ajouter à la suspension du complément
qui détruira les cellules sur lesquelles se sont fixées les anticorps.
C – 4 – Cluster de différenciation
La conclusion
principale de l’étude des sous populations lymphocytaires est que des
lymphocytes exprimant des combinaisons particulières de protéines membranaires
représentent en fait soit des cellules à des étapes de différenciation
différente, soit des cellules ayant des propriétés fonctionnelles différentes.
Pour ces raisons, ces molécules de surface ont été appelées antigènes de
différenciation. Lorsqu’un groupe d’anticorps monoclonaux permet de reconnaître
le même antigène de différenciation, on dit qu’ils définissent des clusters de
différenciation et sont noté CD suivit d’un nombre définit de façon arbitraire.
Ces clusters de différenciation sont à l’origine de la nomenclature actuelle
des molécules de surface permettant de distinguer les différentes sous
populations lymphocytaires.
D – Etude
fonctionnelle des sous populations lymphocytaires
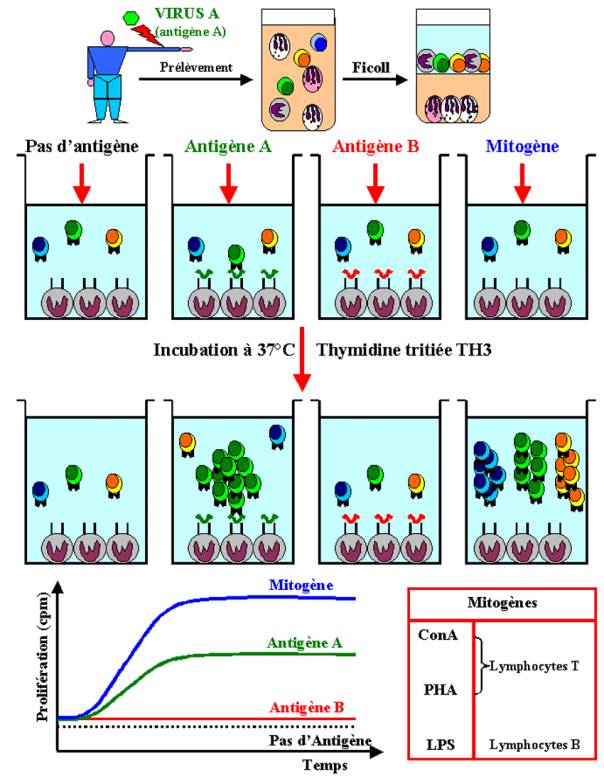
D – 1 – Réaction de prolifération lymphocytaire
Pour permettre une
immunité adaptative, après stimulation par un antigène, les rares lymphocytes
spécifique de cet antigène présents dans l’organisme doivent proliférer
intensément avant de se différencier en cellule effectrice. Cette étape
initiale de prolifération permet de générer assez de cellules effectrices pour
faire face à l’agression antigénique. Le nombre de cellules qui prolifèrent à
la suite d’une stimulation antigénique étant relativement faible et donc
difficilement détectable, le système de mesure doit d’abord être calibré en
utilisant des mitogènes polyclonaux. Ces substances ont pour propriété
d’induire la prolifération des lymphocytes quelle que soit leur spécificité
antigénique. Certains mitogènes agissent seulement sur les lymphocytes T
(Concavaline A, Phytohemagglutinine), sur les lymphocytes B (LPS) ou
indifféremment sur les deux populations (Pokeweed).
Les mitogènes
polyclonaux induisent les mêmes mécanisme de prolifération que ceux induits par
l’antigène. En effet, à l’état basal, le lymphocyte est une cellule au repos
bloquée en phase G0 du cycle cellulaire. Lorsque ces cellules sont activées
soit spécifiquement par l’antigène, soit de manière polyclonale par les
mitogènes, les cellules passe au stade G1 et progressent ensuite rapidement
dans le cycle cellulaire. Dans la plupart des études, la prolifération
lymphocytaire est mesurée par l’incorporation de thymidine tritiée dans l'ADN
des cellules en division. Ce type de test est couramment utilisé en clinique
humaine pour mesurer si chez les patients ayant une suspicion de déficit
immunitaire, les lymphocytes T sont capables de répondre à une stimulation
antigénique spécifique ou non.
Lorsque la
culture lymphocytaire a été optimisée en utilisant la réponse proliférative aux
mitogènes polyclonaux, il devient alors possible de détecter la prolifération
des lymphocytes T spécifiques de l’antigène in
vitro en mesurant l’incorporation de thymidine tritiée en réponse à
l’antigène vis à vis duquel les lymphocytes T du donneur ont été préalablement
sensibilisés. Ce test permet l’évaluation de la réponse des lymphocytes T de
type CD4+ auxiliaires. Cette méthode est un test global qui ne renseigne en
rien sur les capacités fonctionnelles des cellules répondeuses.
D
– 2 – Mesure des fonctions effectrices des lymphocytes T
Les cellules
effectrices sont détectées par les effets qu’elles induisent sur des cellules
cibles présentant des peptides antigéniques spécifique à leur surface ou par la
sécrétion de cytokines particulières agissant spécifiquement sur les cellules
cibles. La mesure de ces fonctions effectrices est à la base de tests
biologiques permettant d’évaluer aussi bien la spécificité du lymphocyte T pour
l’antigène que l’activation des fonctions effectrices de la cellule T.
D
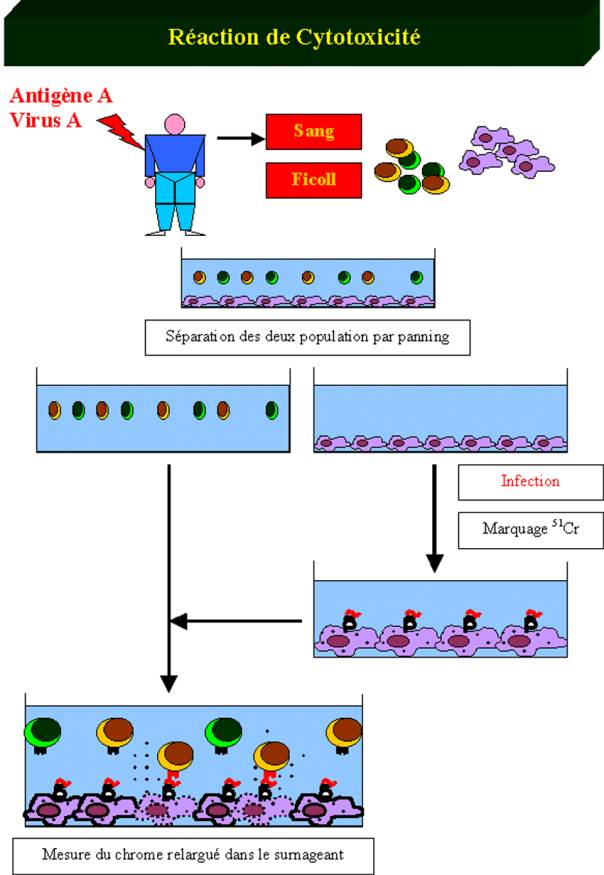
– 2 – 1 – Réaction de cytotoxicité cellulaire
Les lymphocytes
T CD8+ activés tuent toute cellule qui exprime à sa surface des complexes CMH
de classe I/peptides susceptibles d’être reconnus par le récepteur de
l’antigène du lymphocyte T (TcR). Ainsi, la fonction des lymphocytes T CD8+
peut être déterminée en utilisant un simple test de cytotoxicité in vitro. Dans ce test, des cellules
cibles, généralement des macrophages syngéniques sont incubés avec l’antigène
contre lequel ont souhaite déterminer l’intensité de la réponse cytotoxique
essentiellement médiée par les lymphocytes T cytotoxiques CD8+. Le macrophage
exprime alors à sa surface des complexes CMH de classe I/peptides spécifiques
du TcR porté par le lymphocyte T CD8+. Les macrophages sont alors incubés avec
du Chrome 51. Ce traceur radioactif diffuse dans les cellules vivantes et y
reste emprisonné. Les cellules cibles ainsi préparées sont mise au contact de
la suspension cellulaire contenant les cellules T CD8+ effectrices. Si la
suspension contient des lymphocytes T cytotoxiques dont le TcR est spécifique
du complexe CMH de classe I/peptide, ces cellules vont s’activer et détruire la
cellule cible. La cellule cible libère alors en mourant le Chrome 51 dans le
milieu de culture. Le dosage de ce radioélément reflète donc les fonctions
effectrices des lymphocytes T cytotoxiques.
D – 2 – 2 – Mesure des cytokines produites par les
sous-populations lymphocytaires
Les fonctions
des cellules T CD4+ impliquent plus des phénomènes d’activation que des
phénomènes de cytotoxicité à l’encontre de cellules cibles porteuses de
complexes CMH de classe II/peptide spécifique. Lorsque les lymphocytes T CD4+
rencontrent à la surface d’une CPA un complexe CMH de classe II/peptide, le
lymphocyte s’active et prolifère. Cette prolifération peut être mesurée par
l’incorporation de thymidine tritiée. Après cette étape de prolifération, vient
une étape de différenciation. Schématiquement, les lymphocytes T CD4+ peuvent
se différencier en cellules Th1 ou Th2. La différence entre les deux population
cellulaire vient du profil de cytokines sécrétée par ces deux types de
cellules. Les lymphocytes T CD4+ de type Th1 produisent de l’IL-2 et de
l’interféron-g alors que les lymphocytes T CD4+ de type Th2 produisent de
l’IL-4, de l’IL-5, de l’IL-10 et de l’IL-13. Ces cytokines vont exercer leurs
effets sur d’autres types cellulaires. Ainsi, les cytokines Th1 sont de
puissants inducteurs de la réponse à médiation cellulaire et vont notamment
induire l’activation des macrophages et des lymphocytes T CD8+, alors que les
cytokines Th2 vont plutôt stimuler l’immunité à médiation humorale en
favorisant l’activation et la différenciation des lymphocytes B. Ainsi, la
mesure des cytokines produites au cours de la réponse lymphocytaire T CD4+
renseigne sur le type de réponse immunitaire induite après la stimulation
antigénique.
D – 2 – 2 – 1 – Méthodes biologiques
Les cytokines peuvent
être détectées par leur activité biologique sur des cellules dont la
prolifération dépend de la présence de ces médiateurs solubles. Ces test n’ont
plus qu’un interêt historique.
D – 2 – 2 – 2 – Méthodes Immunochimique
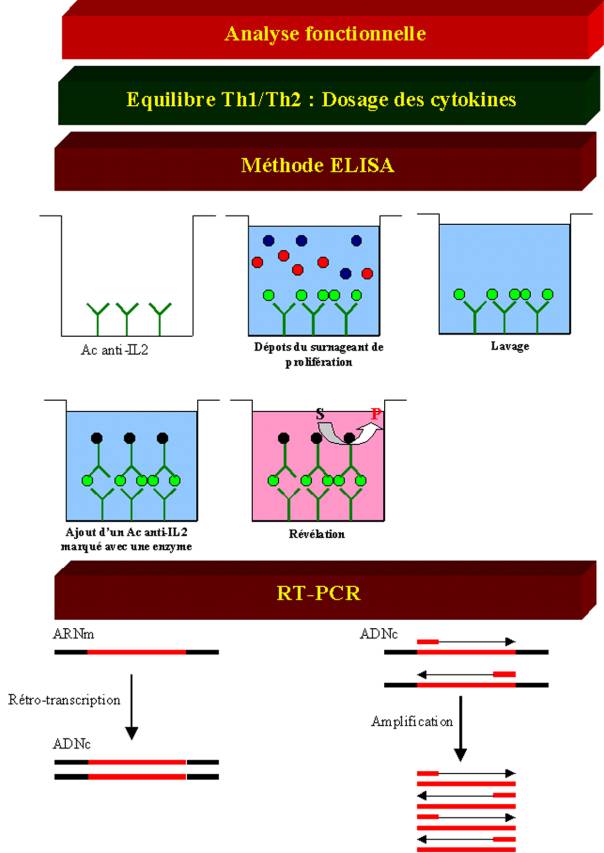
Le test le plus
spécifique et le plus couramment utilisé pour doser les cytokines dérive de la
méthode ELISA et est appelé ELISA sandwisch. Dans cette technique, un premier
anticorps spécifique de la cytokine à doser est fixé au fond d’un puits en
plastique. Cet anticorps est appelé anticorps de capture. Le surnageant de la
culture lymphocytaire contenant la cytokine à doser est alors ajouté dans le
puits. La cytokine se fixe alors sur l’anticorps. Après plusieurs lavages à
l’aide d’une solution saline, un deuxième anticorps lui aussi spécifique de la
cytokine mais reconnaissant un autre épitope sur la molécule est ajouté. Cet
anticorps est marqué par une enzyme et est appelé anticorps de révélation. Si
la cytokine est présente, elle va former un pont entre les deux anticorps,
l’ensemble formant un complexe stable accroché au fond de la plaque. La détection
de ce complexe et donc la présence de cytokines dans le milieu de culture sera
révélée en ajoutant dans le puits le substrat de l’enzyme fixée sur l’anticorps
de révélation, substrat qui sera alors converti en un produit coloré.
Une alternative
à cette technique consiste non plus à détecter la cytokine elle-même mais à
mesurer le nombre de cellules produisant cette protéine. Cette méthode est
appelée ELISPOT. Dans cette technique, ce sont les cellules elles-mêmes que
l’on dépose dans le puits recouvert d’un anticorps spécifique de la cytokine à
doser. Après une courte période d’incubation, les cytokines produites par la
cellule sont captées par les anticorps au contact de la cellule. Après lavage,
on incube la plaque avec l’anticorps de révélation et on ajoute le substrat. La
production de cytokine est détectée par des petites taches au fond du puits. La
numération de ces taches rapportée au nombre de lymphocytes T déposé dans le
puits indique le % de cellules produisant cette cytokine. Enfin, assez récemment
est apparue une technique permettant de détecter les cytokines produites par
les cellules T en utilisant la cytométrie en flux. Les cellules à analyser sont
fixées puis perméabilisées de façon transitoire à l’aide d’une solution de
Saponine. Un anticorps spécifique de la cytokine et marqué par un fluorochrome
est incubé avec la suspension de cellules perméabilisées. Les pores
membranaires permettent aux anticorps de diffuser dans la cellule et de se
fixer sur la cytokine. Après un lavage dans un tampon ne contenant pas de
Saponine, les pores de la membrane plasmique se referment emprisonnant les
complexes cytokine/anticorps fluorescent. Il ne reste plus alors qu’à analyser
la fluorescence des cellules par cytométrie en flux pour connaître le % de cellules
produisant la cytokine.
D – 2 – 2 – 1 – Méthodes Moléculaires
Une approche un
peu différente pour mesurer la production de cytokine par une cellule ou un
tissu consiste à déterminer la présence et le niveau d’expression des ARN
messagers codant cette cytokine. Ceci peut être réalisé par hybridation in situ ou par Northern blot. Toutefois,
ces techniques s’avèrent peu sensibles. Un des moyens permettant d’augmenter la
sensibilité de détection des ARN messagers codant une protéine consiste à
utiliser la méthode de Reverse Transcriptase -Polymérase Chain Réaction
(RT-PCR). La Reverse Transcriptase est une enzyme utilisée par certains virus à
ARN pour convertir l’ARN viral en ADN complémentaire. Dans la méthode de
RT-PCR, les ARN messagers sont convertis en ADNc par la reverse transcriptase.
L’ ADNc choisit est alors amplifié sélectivement par réaction de polymérisation
en chaîne en utilisant un couple d’amorces spécifiques. Lorsque les produits de
la réaction d’amplification sont analysés par électrophorèse en gel
d’agarose/BET, l’ADN amplifié peut être visualisé comme une bande de taille
déterminée. La quantité d’ADN amplifié est proportionnelle à la quantité d’ARN
messager présent au départ. La quantité des ARN messagers de cytokine est
généralement déterminée en comparant l’intensité de la bande obtenue avec celle
obtenue après amplification, dans les mêmes conditions, d’un gène
« rapporteur » dont l’expression est constante dans toutes les
cellules. Des techniques plus quantitatives utilisant un plasmide compétiteur
(PCR compétitive), des amorces fluorescentes (procédé TaqMan ou Lightcycler) ou
des PCR-ELISA sont actuellement en cours de développement.